-
Controlling Symmetry-Breaking Charge Separation in Pyrene Bichromophores
J. Wega, K.-F. Zhang, J. Lacour and E. Vauthey
Journal of Physical Chemistry Letters, 15 (2024), p2834-2840


DOI:10.1021/acs.jpclett.4c00337 | unige:175680 | Abstract | Article HTML | Article PDF | Supporting Info

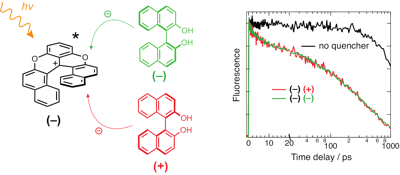
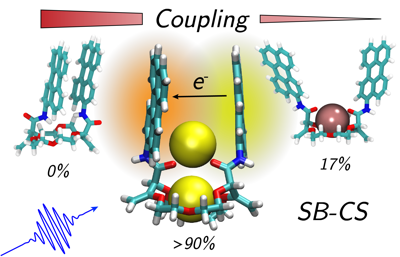
So far, symmetry-breaking charge separation (SB-CS) has been observed with a limited number of chromophores and is usually inhibited by the formation of an excimer. We show here that thanks to fine-tuning of the interchromophore coupling via structural control, SB-CS can be operative with pyrene, despite its high propensity to form an excimer. This is realized with a bichromophoric system consisting of two pyrenes attached to a crown ether macrocycle, which can bind cations of different sizes. By combining stationary and time-resolved spectroscopy together with molecular dynamics simulations, we demonstrate that the excited-state dynamics can be totally changed depending on the binding cation. Whereas strong coupling leads to rapid excimer formation, too weak coupling results in noninteracting chromophores. However, intermediate coupling, achieved upon binding of Mg2+, allows for SB-CS to be operative.